The Price of Not Being Cancer¶
The title of the oldest human being in recorded history currently belongs to Jeanne Calment, who lived to 122. This record remained uncontested for quite a while, with the runner-up, Kane Tanaka, being 3 full years younger. Soon, however, Jeanne Calment's record will become somewhat more controversial (even more than it already is). Henrietta Lacks's cervical cancer cells (HeLa), taken in 1951 when she was 31, are still dividing in labs worldwide; in 2043, this cell line will have "lived" for 122 years, matching Calment's record, with no signs of stopping. Guinness Book of World Records will probably have to take an explicit stance on whether to exclude contenders with unicellular body plans.
There's a temptation to dismiss HeLa's immortality as an artifact of in vitro culture – cells coddled in nutrient broth, a lab peculiarity. But outliving host organisms is well within the capability of mammalian cell lines in the wild. One example is the Tasmanian Devil Facial Tumor Disease (DFTD): a parasitic cancer cell line has been spreading between Tasmanian devils since at least 1986. Devils themselves live only 7 years max; the DFTD cell lineage has already far outstripped that.
Still, one might argue that DFTD cells are a product of disease, an aggressive pathology, perhaps evolutionarily unstable on a scale of the human lifespan. Haha, no. Canine Transmissible Venereal Tumor (CTVT) is a lineage of dog cancer cells, transmitted between dogs during mating. Current estimates place CTVT cell line origin 11,000 years ago. That means at the very beginning of the Neolithic revolution, one dog developed a venereal tumor, and its cells have been clonally propagating across the globe ever since, through skin-to-skin contact. All the other cells of that original dog are long dead, but CTVT is still with us, rawdogging through the entire Holocene since the origin of written language. Good boy.
The reason we don't see any older transmissible tumors is not well established, but it seems plausible that over sufficiently vast timescales these cell lines do finally succumb to Muller's Ratchet – as would any purely asexual replicator. For our purposes though, an 11,000-year run counts as "basically biologically immortal" at the cellular level. As a cellular lineage, CTVT is an undeniable evolutionary success story.
(And yes, fair warning: I'm about to anthropomorphize evolution more than a Pixar movie. If that bothers you, mentally replace every "evolution decided" with "selective pressures resulted in." We both know what's really happening.)
The machinery for such indefinite replication isn't some alien technology; it's latent within our own cells. Immortalizing normal human cells in the lab, while requiring intervention, often involves just tweaking a few known pathways – p53, Rb, telomerase. It's almost insulting how simple it can be.
If cellular immortality is biologically achievable, and demonstrably a long-term winning strategy for cell lineages like CTVT in the wild, why isn't this the norm for all our somatic tissues? If the CTVT lineage can thrive for millennia, why do the cooperative cells that constitute a dog's body, or a human's, senesce and fail within mere decades? If the "software" for immortality is present in our cells, why haven't our normal tissues evolved to routinely deploy it for the benefit of the whole organism, leading to vastly extended lifespans? Why aren't dogs living for 10,000 years then? There must be a catastrophic downside for the multicellular organism that outweighs the cellular benefit of immortality. What is it?
II.
Let's talk about what tumor cells actually do:
-
Multiply blazingly fast
-
Don't die when told
-
Hog resources
-
Ignore nearby cells
-
Evolve to get better at all the above
Sorry, did I say "tumor cells"? My bad, "single cells". It's what living unicellular organisms do.
For billions of years, this was life: unicellular organisms compete; mutations fuel adaptation; lineages rise and fall. That's the baseline. Multicellularity is a hard-won, highly regulated truce imposed upon this malthusian free-for-all. When a cell 'becomes cancerous,' it's systematically dismantling that truce, effectively 'reverting' to the ancient playbook of unicellular competition.
If reverting to the ancestral state makes cells immortal, why did organisms evolve away from it? The notion of wholes wearing out 1000x faster than parts is counterintuitive. It's like a boat made of wooden planks rotting in a day, while each plank, pried out, could last years in water.
| Hallmark of aging | Dog cell | CTVT cell |
| Genome | Unstable in 30 years | Still working after 10,000 years |
| Epigenome | Unstable in 30 years | Still working after 10,000 years |
| Telomeres | Attrition in 30 years | Still working after 10,000 years |
| Proteostasis | Lost in 30 years | Still working after 10,000 years |
| Nutrient-sensing | Deregulates in 30 years | Still working after 10,000 years |
| Mitochondria | Dysfunction in 30 years | Still working after 10,000 years |
| Cells | Senescent in 30 years | Still working after 10,000 years |
| Stem cells | Exhausted in 30 years | Still working after 10,000 years |
| Intercellular communication | Altered in 30 years | Still working after 10,000 years |
An alien studying only a HeLa cell might conclude biological immortality is trivial. The alien would be wrong about multicellular organisms, but not about this: foundational cellular hardware supports indefinite operation. Cellular immortality is biologically possible. Why is it restricted to cancer in the wild? Why haven't complex organisms evolved to use this for self-repair and longevity? What's optimal for parts often kills the whole. What keeps the whole alive often kills parts. Aging is not cellular deterioration. Aging is suppressing cellular immortality. Multicellular life creates an internal evolutionary arena: evolution between organisms, and evolution between cells within organisms. This internal pressure isn't just mammalian. Any complex multicellular lineage (sponges, colonial Volvox with its regA gene suppressing "cheater" cells, redwoods) had to evolve mechanisms to stop internal reversion to unicellular competition. Bacterial mats use quorum sensing for collective behavior, hinting at ancient roots of controlling individual replication. Specific solutions and their costs vary.
Part II: How Evolution Tried to Stop Cells From Evolving
Multicellularity establishes a vast internal "ecosystem" of cellular agents. The fundamental challenge isn't just sticking them together; it's preventing this internal ecosystem from immediately collapsing into a Darwinian free-for-all where the most aggressively selfish cell lineages outcompete the cooperative ones, ultimately destroying the host organism. This "reversion to the ancient playbook" of unicellular competition is the constant threat. The causal chain for this threat is straightforward:
-
The Scale of Opportunity (Sheer Numbers): Trillions of cells (
~10^13-10^14in humans), many dividing, create a vast search space for mutations (Total Mutations ≈ D_total_lifetime * μ * GenomeSize). -
The Nature of Somatic Mutation (Seeds of Rebellion): Some mutations (
m_c) will inevitably confer a local selective advantage (faster division, resistance to controls, etc.). -
The Power of Exponential Growth (Cheaters Prosper Locally): A cell with
m_c(dividing atr_cheater > r_normal) sees its lineage expand exponentially:N_cheater(t) / N_normal(t) = e^((r_cheater - r_normal)t). -
The Multi-Hit Ratchet (Evolution of a Monster): Expanding "cheater" clones provide more targets for further advantageous mutations, leading to the accumulation of multiple "hits" (typically 3-7 driver mutations) for full cancer.
This internal Darwinian pressure necessitates powerful, multi-layered defenses. Each layer attempts to counter specific "cheating strategies" that arise as cells try to dismantle the multicellular truce.
Layer 0: Foundational Fidelity – The "Replicative Credit" Printing Press & Quality Control The baseline error rate of DNA replication (μ_raw) and ongoing DNA damage constantly generate mutations, the raw material for any subsequent cellular "jailbreak." If this rate (P(m_c) per division) is too high, the system is overwhelmed with potential defectors from the start. Evolution's first line of defense, therefore, was to invest heavily in High-Fidelity DNA Replication & Repair Machinery. This includes proofreading polymerases and complex repair pathways (Mismatch Repair, Base/Nucleotide Excision Repair, Double-Strand Break Repair). These systems reduce the effective mutation rate to μ_repaired (where μ_repaired << μ_raw). A lower μ_repaired means fewer dangerous mutations accumulate per N total cell divisions, effectively increasing the total pool of "safe" replicative credits the organism can spend before cancer risk becomes too high. This is the foundation of its "replicative credit" budget. Organisms like flatworms or Hydra, with extreme regeneration from pluripotent stem cells (neoblasts), must possess exceptionally high Layer 0 fidelity in these neoblasts to support their strategy of continuous renewal from a large budget of high-quality replicative credits. The price for this Layer 0 fidelity is the energetic cost of these complex repair systems, and they are not perfect; mutations still occur, necessitating further layers.
Layer 1: Establishing the 'Principal' – Germline Supremacy, turning public goods into club goods.
Clonal Reset, and Somatic Subordination True multicellularity, especially in animals, required creating a "principal" class of cells (the germline) whose interests (long-term lineage propagation) are paramount, and a vast "agent" class (the soma) subordinated to serve it. A key problem is preventing somatic mutations from becoming heritable. If all cells could contribute to the next generation, any somatic mutation giving a cell a replicative advantage (even if harmful to the organism) could be passed on, leading to rapid degradation of organismal integrity. The defense is Germline/Soma Separation: early segregation of a protected germline. All other cells form the "disposable" soma. Consequently, the probability of a somatic mutation entering the germline (P(somatic_mutation_enters_germline)) becomes extremely low. Somatic evolution is an evolutionary dead end for the organism's lineage. The trade-off is that the soma accumulates damage not "cleansed" by selection in the same way, contributing to somatic aging, and beneficial somatic adaptations are lost. Further securing this is The Weismann Barrier. Even with initial germline/soma separation, a somatic cell might "dedifferentiate" and contribute to the germline. Molecular and developmental mechanisms (often epigenetic) in many animals make this very difficult. This further drastically reduces P(somatic_mutation_enters_germline). The price is reduced regenerative potential compared to organisms like many plants, which lack a strict Weismann barrier and can often regenerate germ cells from somatic tissues (plants, of course, just laugh at the Weismann barrier and grow new bits from wherever they please. Anarchists, the lot of them). Another challenge is the transmission of accumulated somatic mutations if organisms reproduced via multicellular propagules. If an organism started from 10 cells instead of 1, and any of those carried "cheating" mutations, the new organism could be compromised from the start, or cheaters would have a head start. The defense is the Unicellular Bottleneck (Zygote): starting each new sexual generation from a single cell. This is a "clonal reset," purging almost all prior somatic variation. Any mutation in the zygote faces intense early developmental selection; P(system_viability | severe_early_cheat_mutation_in_zygote) is very low. The price is the extreme vulnerability of this single-cell stage. (Flatworms using fission manage somatic mutation purging differently, likely via their neoblast system). Finally, to create a vast, functional soma where most cells are specialized and less "dangerous," rather than a democracy of equally competitive cells, Asymmetric Division in stem/progenitor cells is employed. One daughter cell remains a stem/progenitor, the other commits to differentiation with limited subsequent divisions. This establishes a hierarchy. Most somatic cells are "born" with limited "replicative credits" and a restricted fate. P(somatic_cell_becomes_rogue_stem_cell) is kept very low. The price is limited tissue plasticity and repair capacity.
Layer 2: Rationing "Replicative Credits" & Controlling Somatic Proliferation – The Pareto Front of Size, Longevity, and Regeneration Even with a subordinated soma and high-quality "replicative credits" from Layer 0, somatic cell divisions create opportunities for "cheaters." Layer 2 manages how the overall replicative credit budget (determined by Layer 0, controlled by Layer 1) is spent across body size (N_system_agents), lifespan (T_lifespan), and regenerative capacity (R_regen). These form a Pareto front: maximizing all three simultaneously is typically not possible with a finite budget of "safe" cell divisions. Trying to "overextend" this front without a larger credit budget or more robust defenses leads to cancer. One set of cellular cheating strategies involves "Shredding the Social Contract (Loss of Growth Control)": normal cells wait for 'permission slips' (growth factors) and respect 'no vacancy' signs (contact inhibition). A cancer-initiating cell hotwires its growth factor receptors or disables internal brakes (e.g., RB mutations), stopping its listening to the collective. The organism's defense is Strict Growth Controls (Growth Factor Dependence, Contact Inhibition), tying replication to external signals and density. This keeps r_cell coupled to organismal needs, minimizing (r_cheater - r_normal). The price is slower wound healing if signals are suboptimal and creating dependencies cancer can exploit. Another cheating strategy is "Claiming Immortality (Reactivating Telomerase)": most somatic cells have a divisional limit via telomere shortening. Cancer cells reactivate telomerase for limitless replication. The organism's defense is Telomere Shortening (Imposing D_max_lineage), acting as a divisional counter. This is a key solution to Peto's Paradox (why large/long-lived animals don't have proportionally higher cancer rates). Organisms tune D_max_lineage based on Layer 0 fidelity, N_system_agents, and T_lifespan. * Large, Long-Lived (Whales, Elephants): Must have stringent D_max_lineage (shorter telomeres/faster attrition) and excellent Layer 0 fidelity and robust Layer 3 policing. Each lineage gets few "credits." Overextending (e.g., whale size with mouse telomere strategy) means catastrophic cancer rates. The price is reduced regenerative capacity. * Small, Short-Lived (Mice): Can "afford" larger D_max_lineage. They "spend" their smaller credit budget lavishly per lineage. Overextending (e.g., trying for extreme longevity without changing this) would likely hit cancer limits. Their short lifespan means they "fall short" of theoretical maximum longevity if cancer were their only constraint. * Humans (Intermediate): Our Hayflick limit reflects our balance. * Conceptual "Math": T_cancer_free ≈ 1 / (N_system_agents * R_cancer_emergence_per_lineage). R_cancer_emergence_per_lineage depends on Layer 0's μ_repaired, k (hits needed), and Layer 2's D_max_lineage. * Life History Extremes: Octopus (short-lived, semelparous) spends credits on rapid growth. Social Insect Queens (long-lived) suggest exceptional Layer 0/unique maintenance. Plants use different strategies (modularity, totipotency). Operating "off" the evolved Pareto front is risky. Overextending leads to increased cancer. Falling short means unrealized somatic durability. Martincorena studies show "cheater" lineages constantly test these limits.

Layer 3: Active Policing (Fine, We'll Hunt You Down) – Eliminating Defector Agents When "cheaters" bypass Layers 0-2, the game shifts to active detection and elimination. One set of cellular cheating strategies is "Ignoring Deportation Orders (Evading Apoptosis)": cells with significant damage are supposed to self-destruct. Cancer cells disable these pathways (e.g., crippling p53) and refuse to die. The organism's defense is The p53 Network (Apoptosis & Cell Cycle Arrest), the "Guardian of the Genome," detecting damage/oncogenic signals and triggering arrest or apoptosis. This aims for a high P(apoptosis | damage_signal > Threshold). The price is loss of potentially repairable cells and strong selection for p53 mutations in cancer. Another set of cheating strategies involves early "Seceding from the Union (Becoming Self-Sufficient & Invasive)": cells displaying "altered-self" markers, potentially evading local controls or probing boundaries. The organism's defense is Immune Surveillance, with patrolling immune cells (NK, CTLs) recognizing and eliminating these cells. The goal is a high P(elimination_of_clone_C_by_time_t). Even sponges show ancient roots of this with amoebocytes. The price is energetic cost, autoimmunity risk, and cancer evolving immune evasion. Further "seceding" involves localized "cheater" clones spreading, invading, inducing angiogenesis, and metastasizing. The organism's defense is Physical Compartmentalization (ECM Barriers, Tissue Architecture), with basement membranes and connective tissue creating barriers. This reduces P(metastasis | local_tumor_of_size_X). The price is limited nutrient diffusion and fibrosis impairing organ function.
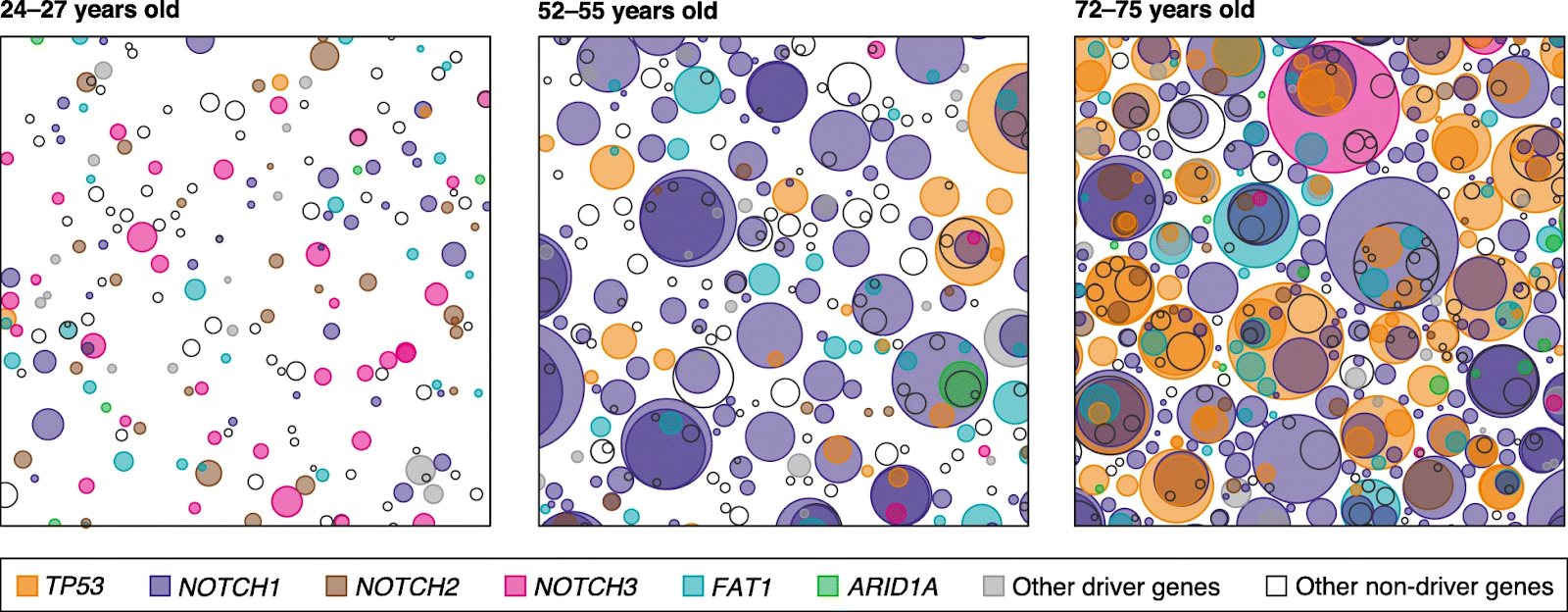
In adult organisms, many cells have mutations that allow them to multiply faster than their neighbors. . In one study, by middle age, more than half of the esophageal epithelium was colonized by mutant clones. In another study, every square centimeter of adult skin was found to have hundreds of cells with pro-proliferation mutations.
The cancer incidence curve is the ultimate "mathematical" scorecard. It shows that over time, P(at_least_one_lineage_wins_defection_game) increases. Organismal lifespan is tied to how long defenses keep this probability low.
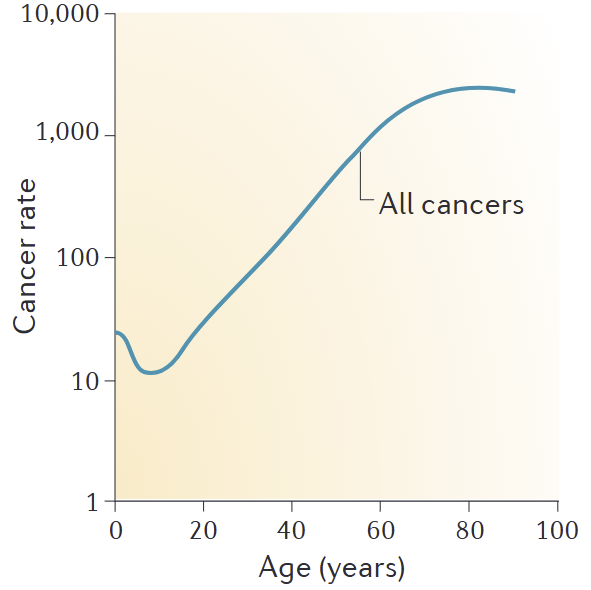
Image: Cancer incidence per 100,000 people in US, data from 1999–2009. Source. So what does an organism do when it's losing the arms race against its own cellular immortality?
Part III: When All Else Fails, Shut Everything Down (a.k.a. Aging)
A common, and quite reasonable, starting point for thinking about why we age goes something like this: evolution's main job is to get our genes into the next generation. Once that's largely accomplished, the selective pressure to keep us in perfect working order drops off a cliff. If a gene helps you reproduce effectively but has deleterious effects that only manifest when you're 70, natural selection, having already seen its primary objective met, isn't going to be very motivated to weed that gene out. This is the core of the "selection shadow" idea. It suggests that aging is what happens in that shadow – a period where the body simply isn't optimized for indefinite survival because there's no strong evolutionary return on that investment. Things wear out, repair systems become less efficient, and damage accumulates, not because of a "program for decline," but because of a lack of a program for perfect, eternal maintenance.
And this makes a lot of sense. It explains why organisms aren't immortal. If this were the entire story, however, we might expect aging to look primarily like a process of passive decay – systems gradually failing, like an old machine left untended, accumulating rust and breakdowns due to simple neglect and the lack of pressure for perfect upkeep. The "check engine" light might come on, but the car itself isn't actively trying to dismantle its own transmission.
Yet, when we scrutinize the actual mechanisms of aging across many species, some of them don't quite fit this picture of mere passive neglect. They can look surprisingly active, conserved, and almost... deliberate. It's not just that the body stops trying so hard to fix things; in some cases, it seems to be actively initiating processes that lead to functional decline. Why would a system that evolution "no longer cares much about" engage in such complex, seemingly counterproductive, and energetically costly shutdown procedures? If it's all just about the warranty expiring, why the elaborate, self-sabotaging rituals?
This is the puzzle that prompts a deeper look. What if some of these active, late-life degradations are not just the result of evolutionary indifference, but are instead the long-term, perhaps overzealous or miscalibrated, consequences of defense systems that were absolutely critical during the reproductive lifespan? (Yes, I know, this 'aging as a grand anti-cancer conspiracy' angle is a bit of a hot take. Your friendly neighborhood gerontologist is probably already drafting a stern letter about good old-fashioned entropy and accumulated damage – and they'd have a point. We're just shining a flashlight on one particularly shadowy corner of the aging labyrinth.)
So what does an organism do when it's losing the arms race against its own cells, when prevention, control, and policing all start to fail? It implements the nuclear option. Here's the terrible question: What if the only way to prevent cancer is to shut down the very processes that keep you alive? I find it best to think of this kind of adaptation as the aging body transitioning into a low-trust, paranoid police state, trading vitality for security. Think about what happens in human societies when trust breaks down. You don't just add more police - you fundamentally change how everything operates. Curfews. Checkpoints. Neighbors reporting neighbors. Everyone becomes a potential threat, so you lock everything down.
That's aging: the final solution to cellular immortality. Not a gentle fading, but a series of increasingly desperate, self-inflicted wounds designed to make the organism an ever-more-barren wasteland for rogue, ambitious cells.
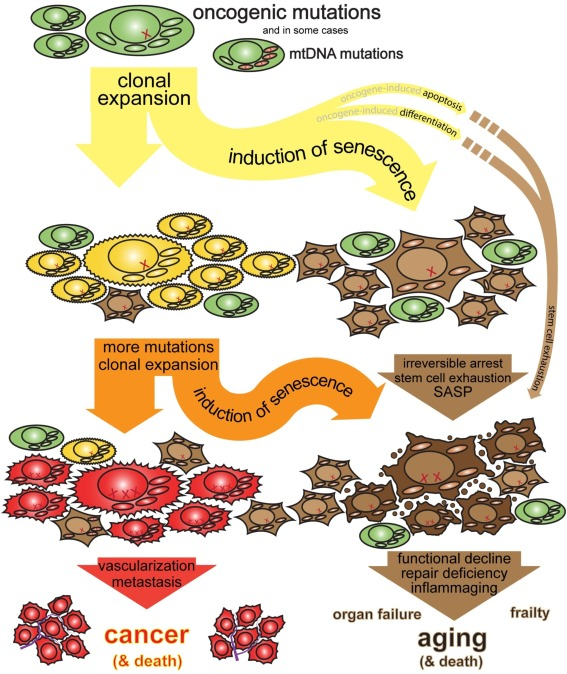
Consider Cellular Senescence. Common wisdom holds that senescence is a good thing: it's an arrest of damaged or stressed cells, important for embryonic development and wound healing by clearing out problematic cells and orchestrating tissue repair. The "anti-cancer lens," however, focuses on why it becomes a driver of aging. When a cell flirts with cancerous transformation, senescence slams on the brakes, permanently halting its division. This is a potent tumor suppression mechanism. The problem is, these arrested "zombie" cells don't just sit quietly; they spew out a nasty cocktail of inflammatory signals and tissue-degrading enzymes (the SASP). In youth, this SASP is a temporary "HELP ME!" signal that recruits immune cells to clear the senescent cell and repair the tissue. But in an aging organism, with a rising tide of pre-cancerous or damaged cells constantly triggering this emergency state, the SASP becomes chronic. The "HELP ME!" signals turn into a constant, damaging background roar. The proportion of senescent cells climbs from near zero to 10-15% in old skin, even 35% in aged primate livers. The anti-cancer benefit is that most dangerously mutated cells do become senescent (the ones that don't can become tumors). The cost of this persistent anti-cancer vigilance is chronic inflammation, tissue degradation, and a microenvironment that perversely can even promote cancer in neighboring cells. It's an emergency brake rusted in the "on" position.
Then there's Stem Cell Exhaustion. Common wisdom sees this as stem cells simply running out of steam, a consequence of wear and tear or hitting their replicative limits. The anti-cancer lens offers a more active interpretation: stem cells, being long-lived and highly proliferative, are prime targets for accumulating oncogenic mutations. So, the body deliberately throttles them down with age – divisions are limited, potency curtailed. This is a powerful tumor suppressive move, reducing the number of "lottery tickets" for cancer initiation in these critical cell populations. While the disposable soma theory also suggests evolution wouldn't invest in eternal stem cell vigor, the active downregulation fits the anti-cancer program narrative. The price is clear: diminished tissue repair, slower wound healing, and fading vitality, all traded for a lower risk of a stem cell turning traitor. Organisms like flatworms or Hydra, which exhibit extreme regeneration and apparent biological immortality, must employ very different strategies to manage cancer risk in their highly active stem cell populations, likely involving continuous cell turnover and stringent quality control that mammals have traded for other advantages.
And where regeneration fails, Fibrosis takes over. Common wisdom views fibrosis as scar tissue formation, a normal response to injury that patches things up. The anti-cancer lens highlights its role as "paving over a rebellious district." When tissue damage is extensive or potentially contains rogue cells, laying down inert scar tissue is a quick, dirty, but effective way to contain the threat and maintain structural integrity. It's a physical barrier against cellular insurrection. But when this becomes the default repair mode in aging tissues – perhaps because the threat of cellular misbehavior is perceived as constant – organs stiffen and lose function. The heart, lungs, liver become battlegrounds where functional tissue is sacrificed for containment. A vital acute defense becomes a chronic, debilitating feature of old age.
Finally, Inflammaging. Common wisdom attributes the chronic, low-grade inflammation of aging to a combination of factors, including immune system dysregulation and responses to accumulated cellular debris. The anti-cancer lens sees inflammaging, in large part, as the immune system stuck on "threat level orange." This sustained vigilance is fueled by the constant emergence of senescent cells spewing SASP, by molecular damage patterns (DAMPs) released from stressed or dying cells, and by the immune system's ongoing surveillance for covertly proliferating mutated cells. While a primed immune response is good for fighting off pathogens (an early-life benefit with late-life inflammatory costs, per antagonistic pleiotropy), its chronic activation due to the internal "cold war" against cancer contributes directly to many age-related diseases, from atherosclerosis to neurodegeneration.
Each of these mechanisms, then, can be seen not just as passive decay or an unfortunate side effect of other evolutionary pressures, but as components of an increasingly aggressive, system-wide anti-cancer strategy. The tragedy is that these defenses, when maintained or escalated in the long run, become major contributors to the very decline they are, in part, trying to prevent the organism from succumbing to (via cancer). The price of suppressing cellular immortality is, in many ways, aging itself.
Looked at this way, aging starts to resemble an active, late-life strategy of doubling down on cancer suppression, accepting widespread functional decline (stiffness, slow healing, reduced reserves) as the necessary price for keeping the internal Darwinian beast caged just a little longer. The Goddess of Everything Else - evolution's cooperative side - comes off as a paranoid, exploitative, nepotistic, totalitarian demiurge with a terminal case of tall poppy syndrome - a patron deity of This Is Why We Can't Have Nice Things. If this view is even partially correct – that much of aging is an active anti-cancer program – it raises serious red flags for simplistic anti-aging strategies. Simply disabling the suppressive mechanisms of aging (like clearing all senescent cells with senolytics, or broadly stimulating growth pathways like mTOR, or, Heaven forbid, extending telomeres) without fixing the underlying reasons they arose (accumulated damage, declining surveillance fidelity) could be like disabling your sprinklers because they damage the carpet, despite keeping lit cigarette butts all over the place. We seem caught in a fundamental bind: we need our cells to be active and adaptable for life, but that very activity allows the possibility of mutation and evolutionary cheating (value drift). Suppressing the cheating hinders function and causes decline (the paranoid mechanisms). It's a constant, exhausting balancing act between tissue degeneration and becoming a 11,000 year old hyper-competitive venereal disease. Is there a way out of this trap? Perhaps the core bargain of multicellularity, this grand project of managing internal evolution, is inherently temporary. Viewing it this way, we exist because of an astonishingly complex system of controls holding back the unicellular tide within us. Cancer is the breakdown of those controls, and aging might be, in large part, the system's final, costly, and ultimately insufficient attempt to reinforce the dam. There is a price to pay for not being cancer, it's paid over decades, and it's what many call aging. We started with a paradox: cells can be immortal, but organisms can't. Now we see why. Aging is not a failure of cellular machinery. Aging is the price of suppressing cellular success. At every scale - from individual mutations to entire organ systems - the pattern repeats: suppress competition, create dysfunction, suppress the dysfunction with more extreme measures, create more dysfunction. It's turtles all the way down, except the turtles are increasingly desperate anti-cancer mechanisms and at the bottom turtle everything just shuts off.
Epilogue: The Uncomfortable Bargain
So, where does this leave us? If this whole "aging as anti-cancer" story holds any water, it paints a rather unsettling picture. It suggests that what we experience as the decline of old age isn't just a passive wearing out, but an active, programmed demolition – a last-ditch, scorched-earth defense against our own cells' relentless drive for immortality. Our bodies, in a sense, are designed to become inhospitable fortresses, sacrificing vitality piece by piece to stave off the internal rebellion that is cancer. The very mechanisms that allow us to live complex, multicellular lives also sow the seeds of our eventual, systemic shutdown. It's less a graceful fading and more a controlled, strategic retreat into oblivion, all to avoid becoming a runaway cellular success story like that 11,000-year-old dog tumor. This isn't to say we should throw our hands up in despair. But it does reframe the challenge of "solving aging" rather dramatically. It's not just about fixing broken parts; it's about renegotiating a fundamental, ancient contract our biology made – a contract where the fine print reads: "Extended multicellular existence requires eventual, systemic self-sabotage." Charming, isn't it?
Thinking about it this way, if one were to, say, look at the burgeoning field of longevity startups with a critical eye (and perhaps a checkbook), this perspective might suggest a few pointed questions to ask, or at least some default assumptions to be wary of.
Takeaways for non-biologists interested to invest in longevity startups:
-
If an "anti-aging" therapeutic claims to extend lifespan by "making cells more resilient" or "making cells immortal", it's best to assume it's a scam. Lack of cellular resilience is likely to be a feature, not a bug. Cells being too resilient is bad! Watch out for buzzwords like "cellular rejuvenation," "resetting the aging clock," or "restoring youthful gene expression" - these are the "quantum" of longevity marketing.
-
Be by default suspicious of any "longevity therapy" that suppresses one or a small number of genes. Evolution preserved these genes for a reason, and as Chesterton's fence tells us, it's best to be very sure what that reason is before getting rid of them. The exception here is for developmentally active oncogenes that should always be silenced in adults anyway - making those oncogenes harder to reactivate in adult cells is probably a good idea.
-
More healthspan doesn't necessarily translate to more lifespan. It's very possible for a therapy to make an organism look healthier in mid-life, and even perform better on health metrics, while at the same time shortening this organism's lifespan. Vigor and frailty aren't necessarily reliable proxies of lifespan. Always demand your anti-aging therapy to show actual lifespan data, not just frailty proxies.
-
Genome stability is best viewed as "replication credits" that the organism can "spend" on either regeneration, size, or longevity - you can't have all three. If a therapy claims to enhance regeneration or its proxies (stem cell proliferation, etc), always demand to see data that show lower mutation rates and increased longevity. Vice versa, even if you have data for increased longevity in some animal model, check for lower regeneration rates and smaller body sizes as likely side effects given constant mutation rate.